Research
Developing innovative experimental and computational methods for causal gene identification in complex diseases and for supporting precision medicine
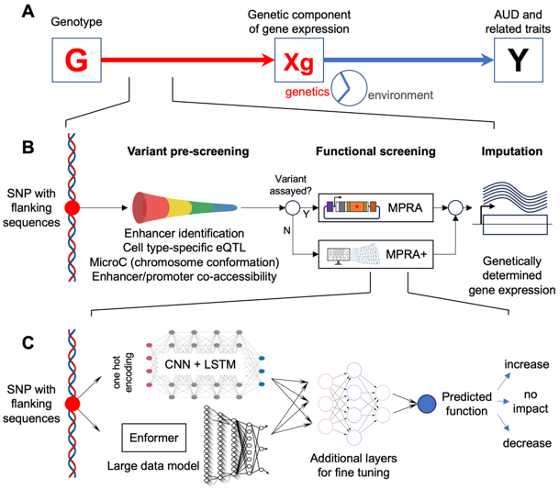
Recent advances in genetics and functional genomics provide exciting opportunities to identify the influence of genetic variation in complex disorders. However, the still limited power of genome-wide association studies (GWAS) for these disorders, combined with the difficulty of identifying functional variants in associated loci, has limited progress. Even when well powered, GWAS identify loci that span many variants, even after follow-up fine mapping studies. This has greatly hampered the identification of functional variants that contribute to the risk for complex diseases, and thereby the understanding of pathways and mechanisms involved in their etiology. A partial solution to this problem is the use of massively parallel reporter assays (MPRA) to systematically profile the activity of large numbers of regulatory variants in cell types that are relevant to a specific disorder. The functional information generated can be used to increase the ability of GWAS to identify relevant variants and to point toward their mechanism of action. We have developed MPRA that simultaneously test the functional activity of over 10,000 variants in the 3’-untranslated regions (3’UTR) and enhancer regions (Frontiers in Genetics, 2018, Alcohol Clin Exp Res., 2020, Molecular Psychiatry, 2021, bioRXiv, 2024). We have also developed a series of AI and machine learning models for identifying causal genes contributing to a specific disorder by combining MPRA-derived functional data with GWAS data. The strategy has been applied to substance use disorder in a funded R01 grant with NIDA and a U10 grant funded by the NIAAA. The method and strategy we have developed can be readily utilized in other disease systems. We are currently developing grant proposals for identifying causal genes in Alzheimer’s disease, and to identify functional somatic mutations in cancer.
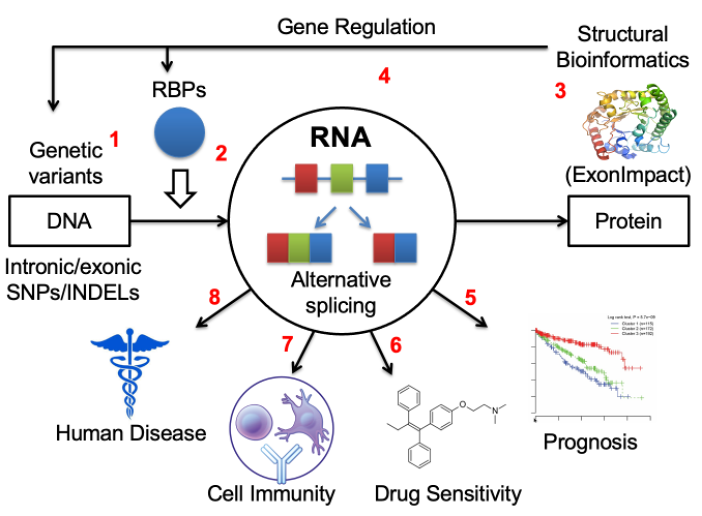
Roles of alternative splicing in complex disease
Over the past 15 years, I have made significant progresses in understanding the splicing regulation and its translational impacts in several aspects, including identify cis-acting RNA elements from CLIP-seq assay by innovatively integrating RNA sequence with secondary structure features (RNAMotifModeler, BMC Genomics 2011), extract novel alternative splicing events from RNA-seq data (Alt Event Finder, BMC Genomics 2012), predicting the pathogenic impacts of the dysregulation of splicing outcomes (ExonImpact, Human Mutation, 2017), discover the roles of exonic and intronic variants on splicing regulation (regSNPs-splicing, Human Genetics, 2017, and regSNPS-intron, Genome Biology, 2019), and examining the correlation between aberrant splicing patterns and cancer patient survival outcomes (Frontiers in Genetics, 2020) as well as the relationships between splicing regulation and drug resistance (Genomics, Proteomics and Bioinformatics, 2022).
Recently, we and others have found that a type of dysregulated RNA splicing events, intron retention (IR), have been commonly observed in tumor transcriptomes. IR occurs when the splicing complex fails to splice introns from the primary messenger RNA transcript and trigger immune responses by either producing disease-specific neopeptides (neoantigens) that can be presented by MHC class I molecules, or generating pseudo-viral response by forming double-stranded RNA molecules. We have published two papers recently concluding that the levels of aberrant intron retention are associated with prognoses in multiple myeloma (Oncogene, 2021) and pancreatic cancer (JCO Clinical Cancer Informatics, 2022). Our preliminary data based on samples collected at IUSCCC precision clinic suggests that patients with higher intron retention levels tend to respond better to the immune checkpoint inhibitor therapy.
In addition to cancer, we recently discovered that aberrant intron retention is also present in other complex diseases such as Alzheimer’s disease, alcohol addiction, alcoholic liver disease, and diabetes. We have also designed a statistical genetics model to identify splicing patterns that contribute to the risk of alcohol use disorder (AUD), by integrating large-scale GWAS data with RNA-seq data derived from post-mortem brain tissues (Molecular Psychiatry, 2023).
Other Research Areas
We have extensive experience in the methodology development and applications on next generations sequencing technology, and computational modeling on transcriptional regulation, microRNA regulation, and epigenetic regulation. Our team has recently invested heavily on developing novel computational methods for single-cell analytics and spatial transcriptomics technologies.